All published articles of this journal are available on ScienceDirect.

Molecular Characterization of HIV-1 Subtypes and Primary Drug Resistance Mutations in Bangladesh: Insights from 2019 Data
Authors Info & Affiliations
Abstract
Introduction
Human Immunodeficiency Virus Type 1 (HIV-1) continues to pose a major public health challenge in Bangladesh, with increasing concern over genetic diversity and emerging drug resistance. This study aimed to investigate the molecular epidemiology, subtype distribution, co-receptor tropism, and Primary Drug Resistance Mutations (PDRMs) in treatment-naïve HIV-1-positive individuals in Bangladesh.
Methods
A cross-sectional study was conducted among 30 treatment-naïve HIV-1-positive individuals enrolled at the Department of Virology, BSMMU. Demographic and geographic data were recorded. RNA was extracted from plasma samples with viral loads ≥2000 copies/mL, and the Reverse Transcriptase (RT) and env gene regions were amplified via nested PCR and sequenced using the 3500Dx Genetic Analyzer. Subtypes were determined using REGA genotyping tools. Phylogenetic relationships were assessed using MEGA-X. Co-receptor usage was predicted via Geno2Pheno, and PDRMs were evaluated using the Stanford HIV Drug Resistance Database.
Results
The study cohort had a male predominance (73%) with a mean age of 37 ± 12 years and originated from all eight administrative divisions of Bangladesh. Half of the participants had a history of international migration, particularly to Saudi Arabia. Subtyping identified all 30 sequences as HIV-1 subtype C using REGA. Phylogenetic analysis revealed strong clustering with regional strains from India, China, Malaysia, Iran, Myanmar, and South Africa, suggesting transnational transmission. Co-receptor analysis showed that 77% of sequences were CCR5-tropic and 23% were CXCR4-tropic. No major NRTI mutations were detected; however, NNRTI resistance mutations (V106I, G190V, V179D) were identified in 13% of cases. Additionally, a high frequency (77%) of the D121 polymorphism was observed.
Discussion
This study provides valuable insights into the molecular epidemiology of HIV-1 in Bangladesh, reaffirming the predominance of subtype C, likely introduced through regional or international migration. The presence of major NNRTI-associated resistance mutations and CXCR4-tropic variants in a portion of treatment-naive individuals, although based on a limited sample size, raises concerns about emerging drug resistance and the potential impact on treatment outcomes. Although no major NRTI resistance mutations were identified, the presence of NNRTI-associated mutations in 13% of treatment-naïve cases is concerning, as transmitted drug resistance can compromise first-line therapy effectiveness. The high frequency of the D121 polymorphism, though not directly linked to treatment failure, reflects underlying genetic variability that may influence future resistance pathways.
Conclusion
This study confirms the predominance of HIV-1 subtype C in Bangladesh. The detection of NNRTI-associated resistance mutations and CXCR4-tropic variants in treatment-naïve cases raises concerns for future treatment effectiveness. This study highlights the need for ongoing molecular surveillance to track subtype diversity, detect early drug resistance, and support evidence-based strategies. Larger, updated studies are essential to validate these results and guide future HIV policies in Bangladesh.
1. INTRODUCTION
Acquired Immunodeficiency Syndrome (AIDS) caused by Human Immunodeficiency Virus (HIV) has been a significant public health concern for over four decades. Globally, more than 75 million individuals have been infected, with an estimated 32.7 million deaths attributed to AIDS-related complications [1]. Even though there are continuous advancements in treatment and prevention, in 2019, about 1.7 million new infections were reported, which represents a 23% decline in new diagnoses compared to 2010 [2].
In Bangladesh, HIV prevalence has remained low in the general population since the first confirmed case in 1989. However, infection rates have reached up to 1% among key vulnerable groups such as Female Sex Workers (FSW), Men Who Have Sex With Men (MSM), Intravenous Drug Users (IDU), and the transgender community (Hijras) [3]. Notably, harm reduction strategies like needle-syringe exchange programs have significantly reduced transmission among IDUs by as much as 90% and helped curtail spread to other populations [4]. However, recent trends show a growing epidemic, particularly among returning migrant workers [5].
Characterization of HIV transmission dynamics is mostly based on molecular epidemiology and phylogenetic analyses, which help to identify public health interventions. HIV-1 remains the predominant strain worldwide, and approximately 56% of infections are accountable for subtype C, followed by subtypes A (23%), B (8%), and various Circulating Recombinant Forms (CRFs) [6]. However, in Bangladesh, limited research has been conducted on the molecular diversity of HIV-1. One of the few large-scale studies analyzing partial gag gene sequences from 198 individuals reported subtype C as the most prevalent (41.4%) [7].
In resource-limited countries like Bangladesh, this is concerning as the emergence of primary Drug-Resistant Mutations (DRMs) can compromise the efficacy of Antiretroviral Therapy (ART), leading to treatment failure, increased morbidity, and higher transmission risk [8]. Regular surveillance and genetic sequencing of HIV-1 strains are critical to monitoring the evolving drug resistance landscape.
Very few sequencing studies have been conducted in Bangladesh in recent years, even though they are urgently required. The latest available data, published in 2020, analyzed sequences obtained in 2016, with drug resistance data limited to only 10 individuals [9]. In this context, the current study aims to bridge this knowledge gap by characterizing the molecular subtypes, co-receptor tropism, and primary drug resistance mutations of HIV-1 among treatment-naïve patients in Bangladesh using updated sequencing and phylogenetic analysis.
2. METHODOLOGY
2.1. Study Design, Participant Selection, Sample Collection, and Viral Load Measurement
This observational study was conducted over one year (January - December 2019) at the Department of Virology, Bangladesh Medical University (BMU), formerly known as Bangabandhu Sheikh Mujib Medical University (BSMMU), Dhaka. A total of 40 purposively selected HIV-1 antibody-positive adults (≥18 years) with confirmed HIV-1 RNA and a viral load ≥2000 copies/ml, and who had not initiated Antiretroviral Therapy (ART), were enrolled. After obtaining written informed consent, socio-demographic and clinical data were collected, and plasma samples were preserved for laboratory analysis. Ethical approval for the study was obtained from the Institutional Review Board of BMU (former BSMMU).
2.2. Viral RNA Extraction
HIV-1 RNA was isolated from 40 stored plasma samples using pathogen free RNA isolation kit from Geneproof (Videnska, Czech Republic) according to the manufacturer's instructions.
2.3. First-strand cDNA Synthesis
For first-strand cDNA synthesis, 5 μl of isolated RNA was taken and then reverse transcribed using the FireScript RT-cDNA synthesis kit from Solisbiodyne using random hexamer (Tartu, Estonia) according to the given protocol.
2.4. Nested PCR Amplification and Gel Electrophoresis of env and pol Gene
For first-round PCR, 5 μl of cDNA product was mixed with 12.5 μl of PCR master mix (Promega, USA), 1.5 μl of each outer-forward and outer-reverse primer, and 4.5 μl of nuclease-free water for a total volume of 25 μl. The reaction mix was then amplified on an Applied Biosystems GeneAmp 2700 thermocycler (Thermo Fisher, USA) using cycling parameters described elsewhere [10, 11]. Both the env and pol gene fragments could be amplified in a total of 30 samples.
2.5. Purification and Sequencing of Amplified Products
Amplified products were then purified, removing excess primers and unincorporated nucleotides using an enzymatic purification method (ExoSAP-IT, Thermo Fisher, USA) according to the manufacturer's protocol. The cycle sequencing reaction was performed using an inner forward primer for each gene and ABI BigDye Terminator 3.1 cycle sequencing kit (Applied Biosystems, USA). Final purification of the cycle sequenced product was performed using BigDye Xterminator purification kit (Applied Biosystems, USA) according to a given protocol. After final purification, products were sequenced in an ABI 3500Dx genetic analyzer (Applied Biosystems, Foster City, USA). Sequences were accepted only if both forward sequencing produced high-quality chromatograms with Phred scores exceeding 20.
2.6. Sequence Analysis and Phylogenetic Tree Construction
Sequence files were manually checked on Chromas 2.3 for quality, insertion, and deletion. Sequences were manually edited in BioEdit version 7.2. Then, sequences were subtyped using the REGA HIV-1 Subtyping Tool Version-3 developed by Stanford University (http://dbpartners.stanford.edu:8080/RegaSubtyping/stanford-hiv/typingtool/). After that, a Multiple Sequence Alignment (MSA) was performed on HIV-1 study sequences and downloaded reference sequences using the native MUSCLE program in MEGA-X with default parameters. Aligned sequences were refined using an online tool, Jalview (https://www.jalview.org/). The phylogenetic analysis of HIV-1 samples was performed in MEGA-X using the Hasegawa-Kishino-Yano model with 1000 bootstrap replicates. The resulting phylogenetic tree was visualized using online software IQ Tree (https://www.hiv.lanl.gov/content/sequence/IQTREE/iqtree.html)
2.7. RT-gene Mutation/Polymorphism Analysis
Primary Drug Resistance Mutation (PDRM) analysis was performed using HIV Drug Resistance Database developed by Stanford University (Available at https://hivdb.stanford.edu/hivdb/by-mutations/). HIV-1 RT-gene mutations were either categorized as Nucleoside Reverse Transcriptase Inhibitor (NRTI)/ Non-Nucleoside Reverse Transcriptase Inhibitor (NNRTI) mutations or minor mutations.
2.8. Statistical Analysiss
All the statistical analyses were performed using SPSS version 23. To observe the association between continuous variables t-test was used, and for categorical variables, a chi-square test was used. P-value less than. 0.05 was reported as statistically significant.
2.9. Ethical Consideration
Written informed consent was obtained from each patient participating in this study. Their personal information was anonymized and stored in a password-protected computer. Ethical permission for this study was obtained from the IRB board of Bangladesh Medical University (BSMMU/2019/7133).
3. RESULTS
3.1. Demographic and Clinical Characteristics
Table 1 describes a total of 30 treatment-naïve HIV-1-positive patients who were enrolled in this study. The cohort had a male predominance, with 22 male patients (73%) and 8 female patients (27%). The overall mean age of participants was 37 ± 12 years, with males having a slightly lower mean age (38 ± 12 years) compared to females (43 ± 13 years).
| Parameter | Category / Details | Number of Patients | Percentage (%) | Mean Age (Years ± SD) | Comments |
|---|---|---|---|---|---|
| Total patients | - | 30 | 100 | 37.6 ± 12.50 | Treatment-naïve HIV-1 positive individuals |
| Gender | Male | 22 | 73.3 | 38.31 ± 12.55 | Majority of patients were male |
| Female | 8 | 26.7 | 42.75 ± 12.60 | ||
| Geographic Distribution | Dhaka | 9 | 30.0 | - | Highest representation |
| Chittagong | 5 | 16.7 | - | ||
| Barishal | 4 | 13.3 | - | ||
| Sylhet | 3 | 10.0 | - | ||
| Rajshahi | 3 | 10.0 | - | ||
| Khulna | 2 | 6.7 | - | ||
| Rangpur | 2 | 6.7 | - | ||
| Mymensingh | 2 | 6.7 | - | ||
| History of Migration | Lived abroad (esp. Saudi Arabia) | 15 | 50.0 | - | Relevant for tracing viral origins |
The patients originated from all eight administrative divisions of Bangladesh, reflecting a wide geographic distribution. The majority of participants were from Dhaka (30%), followed by Chittagong (17%), Barishal (13%), Sylhet and Rajshahi (10% each), and the remaining from Khulna, Rangpur, and Mymensingh (7% each). Notably, 50% of the study participants reported a history of migration abroad, with Saudi Arabia being the most common destination. This highlights the potential for cross-border transmission and viral diversity through regional and international mobility.
3.2. HIV-1 Subtyping and Phylogenetic Analysis
The REGA HIV-1 subtyping tool version 3 classified all samples (100%) as subtype C. Phylogenetic analysis using the Maximum Likelihood method revealed that the majority of Bangladeshi strains showed strong clustering with sequences from India, China, Nepal, and South Africa (Fig. 1). Fourteen clusters with bootstrap support ≥50% were observed. Specifically, the strain BSMMU/HIV/5 clustered closely with an Indian reference sequence (EF469243) with 99% nucleotide similarity and a bootstrap value of 99. Another strain, BSMMU/HIV/22, showed 98% similarity with a Chinese strain (XJN0081), although with a lower bootstrap support (51%). The env gene sequences demonstrated a wide nucleotide similarity range (71–98%), while RT gene sequences were more conserved, ranging from 81–99% similarity, indicating closer genetic relationships, particularly with regional strains from India and China described in Table 2. Regarding the phylogenetic tree with RT gene sequences, five study strains formed a cluster with the Indian reference strain ABO23804, with the highest bootstrap value observed at the distant node. Another large cluster was observed where study strain 27 clustered with Malaysian and Iranian strains on the terminal branch with a bootstrap value of 57. This study strain showed a nucleotide similarity percentage of 88 and 90 with the Malaysian and Iranian strain respectively (Fig. 2). Bangladeshi reference strains clustered among themselves without showing any evolutionary relationship with other reference strains. Overall, on the similarity matrix, percentage identity ranged from 81-99%, with the highest being observed while comparing study strains with the Indian reference strain ABO23804. When both trees are compared, similar patterns are observed for both the env and RT gene, as the study’s strains mostly cluster with other Bangladeshi and Indian strains.

The tree was constructed using 30 study sequences labeled as BSMMU/HIV/1–30 and 54 reference sequences retrieved from GenBank. The reference sequences represent different geographic origins, including Bangladesh, India, Nepal, China, Myanmar, Thailand, Singapore, Saudi Arabia, Iran, South Africa, Australia, the United Kingdom, and the USA. The clustering pattern shows the relationship of the study sequences with globally reported HIV-1 env reference sequences. Phylogenetic reconstruction was performed using the Neighbor-Joining method with bootstrap analysis (1,000 replicates) in MEGA software.
| Parameter | Details / Mutation | Number of Cases | Percentage (%) | Statistical Data | Comments |
|---|---|---|---|---|---|
| HIV-1 Subtyping | REGA: Subtype C | 30 | 100.0 | - | All sequences identified as Subtype C |
| NCBI: Subtype C | 25 | 83.3 | - | Five cases showed discrepancy with REGA tool | |
| NCBI: CRF_07BC | 5 | 16.7 | - | Circulating recombinant forms detected | |
| Phylogenetic Clustering | env gene (≥50% bootstrap) | 14 clusters | 46.7 (of all samples) | - | Strong regional association with India, China |
| Closest matches: Indian strain EF469243 | 1 | 3.3 | 99% similarity, BS=99 | Indicates cross-border transmission | |
| Closest matches: Chinese strain XJN0081 | 1 | 3.3 | 98% similarity, BS=51 | ||
| Co-receptor Tropism | CCR5-tropic | 23 | 76.7 | Median FPR = 74.4% | Typical in early-stage infection |
| CXCR4-tropic | 7 | 23.3 | FPR < 10%; p > 0.05 (age assoc.) | Higher in older patients, not statistically significant | |
| RT Gene Drug Resistance | Major NRTI mutations | 0 | 0.0 | - | None detected |
| Major NNRTI mutations | 4 | 13.3 | - | V106I, G190V, and V179D mutations observed | |
| High-level resistance (G190V) | 1 | 3.3 | - | Associated with efavirenz/nevirapine resistance | |
| Accessory/low-level (V106I, V179D) | 3 | 10.0 | - | May influence therapy in combination | |
| Minor mutations (e.g., D121) | 23 | 76.7 | - | Polymorphic substitution; clinical relevance uncertain |
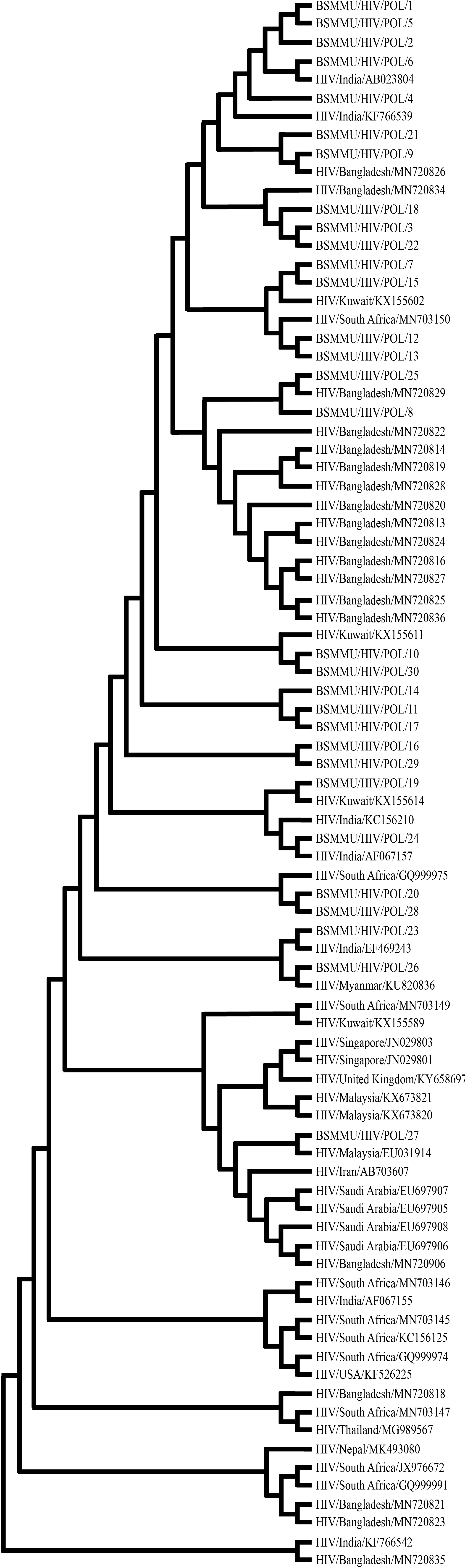
The phylogenetic tree represents the relationship between 30 study sequences (labeled as BSMMU/HIV/POL/1–30) and 55 reference sequences retrieved from GenBank, encompassing multiple geographic regions including Bangladesh, India, South Africa, Kuwait, Saudi Arabia, Iran, Myanmar, Malaysia, Singapore, the United Kingdom, USA, Nepal, and Thailand.
The clustering pattern demonstrates the genetic relatedness of the study isolates with globally circulating HIV-1 strains based on the reverse transcriptase (RT) gene region. Phylogenetic reconstruction was performed using the Neighbor-Joining method with bootstrap analysis (1,000 replicates) in MEGA software.
3.3. Co-receptor Tropism Analysis
Co-receptor usage predictions based on the V3 loop of the env gene showed that the majority of sequences (23 out of 30; 77%) were CCR5-tropic, which is typical of early-stage infections. The remaining 7 sequences (23%) were predicted to use the CXCR4 co-receptor, with False Positive Rates (FPR) below 10%. While there was a trend toward a higher prevalence of CXCR4 tropism among older patients, this association did not reach statistical significance (p > 0.05) in the chi-square test. The median FPR among the study sequences was 74.4%, supporting the predominance of CCR5-tropic viruses in the study population (Table 2).
3.4. Drug Resistance Mutation Profile
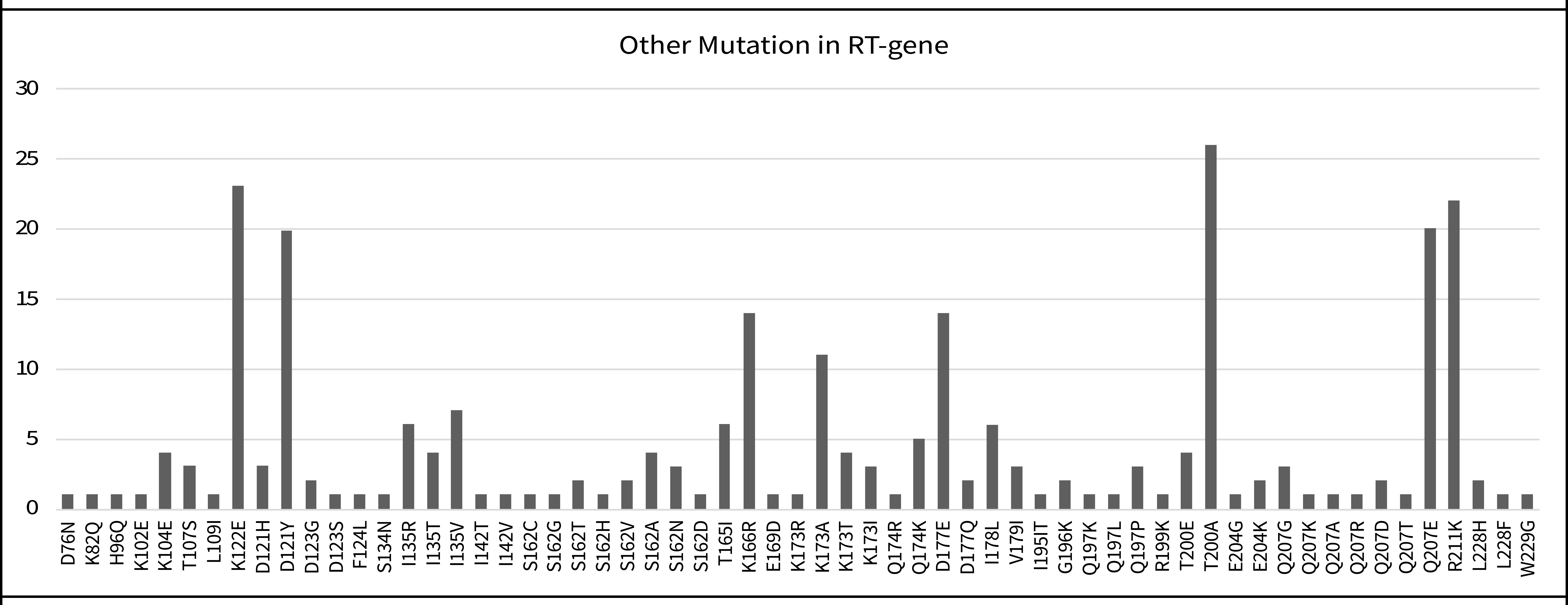
The Reverse Transcriptase (RT) gene sequences were analyzed using the Stanford HIV Drug Resistance Database algorithm to detect the presence of primary resistance-associated mutations. No major Nucleoside Reverse Transcriptase Inhibitor (NRTI) mutations were found among the treatment-naïve individuals, suggesting that first-line NRTI regimens may still be effective in this population. However, four patients (13%) harbored major non-Nucleoside Reverse Transcriptase Inhibitor (NNRTI) mutations. G190V, which was found in one patient (3%), was associated with high-level resistance to both efavirenz and nevirapine. V106I and V179D were found in three additional patients (10.0%), both of which are considered accessory mutations that may contribute to reduced drug susceptibility when present with other mutations. Furthermore, minor polymorphic mutations were observed in a large proportion of sequences. A substitution at position D121 was found in 77% of cases, though the clinical significance of this polymorphism remains uncertain (Table 2; Fig. 3).
4. DISCUSSION
This study highlights the critical importance of integrating HIV-1 molecular surveillance, particularly sequencing-based approaches, into national health strategies to manage and prevent drug resistance in Bangladesh effectively. The study’s comprehensive phylogenetic, tropism, and drug resistance analyses provide crucial insights into the evolving molecular epidemiology of HIV-1 in the country.
HIV-1 subtype C was found in all 30 sequences in the study’s cohort, supporting earlier findings that subtype C is the most common strain in circulation in Bangladesh. [12,13]. This pattern is consistent with the global distribution of subtype C, particularly in Southern Africa and South Asia, where it accounts for the majority of infections [14]. Notably, five sequences (17%) showed similarity with CRF_07BC, a recombinant form more common in East and Southeast Asia, especially China [15]. This finding suggests potential cross-border transmission, most likely due to migration and labor movement, a conclusion bolstered by the fact that 50% of participants had a history of living abroad. Cases of foreign-acquired infections have also been reported in earlier research conducted in Bangladesh, highlighting the contribution of worldwide travel to the entry of various subtypes and recombinant forms into the local epidemic [16].
The phylogenetic clustering of four Bangladeshi sequences within the same clade and their close relationship with sequences from China, India, and Myanmar for the env gene and India, Malaysia, and Iran for the RT gene further supports the hypothesis of regional linkage and transnational transmission routes. This aligns with a study by Neogi et al. [17], which emphasized the genetic closeness of HIV-1 strains across South and Southeast Asia due to migratory flows. Bangladesh’s geographical and economic proximity to HIV endemic countries demands enhanced molecular surveillance at points of entry and within high-risk populations.
In terms of co-receptor usage, the study’s analysis revealed that 77% of the sequences were CCR5-tropic while 23% were CXCR4-tropic, which suggests a dominance of less virulent early-stage infections. This pattern is in line with global trends, where CCR5-tropic viruses are typically more prevalent in newly infected individuals [18]. A previous study by Sarker et al. [19] also reported a majority of CCR tropic viruses among treatment-naive Bangladeshi individuals. However, the detection of CXCR4-tropic strains, nearly a quarter in the study’s cases, is concerning, as these variants are associated with faster disease progression, poorer clinical outcomes, and reduced responsiveness to CCR5 antagonists such as maraviroc [20, 21]. When evaluating the use of the CCR5 antagonist maraviroc, this study highlights demand incorporation of co-receptor tropism testing as a component of customized care. One of the most important contributions of this study is the identification of primary Drug Resistance Mutations (DRMs) in treatment-naïve individuals. In 13% of patients, NNRTI resistance mutations were detected, specifically V106I, G190V, and V179D. These mutations are known to significantly reduce susceptibility to efavirenz and nevirapine, which are widely used first-line Antiretroviral Therapy (ART) drugs in Bangladesh. No NRTI mutations were detected, indicating drug efficacy is maintained for drugs like tenofovir and lamivudine. The 13% prevalence of NNRTI resistance identified in this study reflects a consistent upward trend observed in both earlier and recent studies from Bangladesh, indicating a progressive increase in transmitted resistance among ART-naive individuals [9, 13]. This pattern is further supported by global evidence, which highlights a rising burden of NNRTI resistance, particularly in Low- And Middle-Income Countries (LMICs) [22, 23]. According to the WHO HIV Drug Resistance Report 2021, several countries in sub-Saharan Africa and Southeast Asia have exceeded the 10% threshold for pre-treatment NNRTI resistance, prompting changes in ART guidelines [24]. These findings indicate that Bangladesh is approaching a critical threshold, where treatment outcomes could be jeopardized by the empirical use of NNRTIs without conducting prior resistance testing.
In addition to major DRMs, the current study found that 76% of sequences harbored the D121 polymorphism. Even though they are not directly linked to resistance, such polymorphisms may impact viral fitness or serve as compensatory mutations, facilitating the evolution of high-level resistance when selective pressure from ART is applied [25]. To comprehend the evolutionary potential of HIV-1 in the area, it is imperative to continuously monitor these background mutations. Regular HIV sequencing is imperative for personalized treatment planning, surveilling circulating strains, and creating effective public health initiatives due to the rise of resistance and recombinant forms. Internationally, countries like South Africa and Brazil have incorporated routine genotyping into national HIV programs, resulting in better treatment outcomes and improved drug stewardship [26, 27]. Bangladesh currently lacks such infrastructure outside of research and academic institutions. Establishing a national HIV sequencing program, especially for newly diagnosed and ART-failing patients, would be a significant step toward combating the threat of HIV drug resistance.
Furthermore, the study’s findings provide crucial epidemiological data for policy-makers and reinforce the need to revise national treatment guidelines in light of rising NNRTI resistance. There is a global shift toward dolutegravir-based regimens due to their high barrier to resistance [28]. Bangladesh has begun introducing Integrase Strand Transfer Inhibitors (INSTIs) like dolutegravir in its treatment guidelines, but uptake is still limited [29]. The Integrase region should be included in future sequencing studies to evaluate resistance to more recent medication classes.
5. LIMITATIONS
While this study is among the few in Bangladesh to explore both subtyping and resistance in treatment-naive individuals using dual genomic regions, the relatively small sample size and restriction to partial RT sequences are considerable limitations. 30 samples were sequenced in this exploratory study, which is low compared to other studies. Bangladesh has a low burden of HIV, and hence 30 samples are sufficient to characterize circulating strains, identify dominant subtypes, and identify early events of recombination or transmission clusters. Besides, there was no specific funding for this study either. Also, sanger sequencer was used to sequence the V3 loop region of the envelope gene; the proportion of X4 viruses in samples may have been underestimated, as Sanger sequencing is not as sensitive as next-generation sequencing for tropism determination. Evaluation of mutations in protease or integrase genes was also absent. Future studies should expand coverage to include whole-genome sequencing and representation from all key populations, including PWID, MSM, and female sex workers.
CONCLUSION
This study reinforces the utility of HIV sequencing for drug resistance surveillance and subtyping in Bangladesh. The detection of major NNRTI resistance mutations in treatment-naive individuals, alongside evidence of viral diversity and potential recombination, indicates that drug resistance monitoring should be an urgent priority in national HIV control strategies. Continued investment in sequencing capacity will be critical to preserving treatment efficacy and controlling HIV transmission in Bangladesh.
AUTHORS’ CONTRIBUTIONS
The authors confirm their contribution to the paper as follows: S.U.M.: Conceptualized the study; S.U.M., M.A.M., and N.S.: The methodology was developed; M.A.M., M.Y., T.N., N.S., and J. A.D.: Data curation and formal analysis were performed; M.A.M: Prepared the first draft of the manuscript; S.U.M., S.T., and R.R.: Review and editing of the manuscript were carried out; S.U. M.: Supervised the study. All authors reviewed and approved the final version of the manuscript prior to submission.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Ethical permission for this study was obtained from the IRB board of Bangladesh Medical University (BSMMU/2019/7133).
HUMAN AND ANIMAL RIGHTS
All procedures performed in studies involving human participants were in accordance with the ethical standards of institutional and/or research committees and with the 1975 Declaration of Helsinki, as revised in 2013.
CONSENT FOR PUBLICATION
Written informed consent was obtained from each patient participating in this study.
AVAILABILITY OF DATA AND MATERIALS
All sequence data used in writing up the manuscript are publicly available in the Genbank database.
ACKNOWLEDGEMENTS
The authors would like to pay their gratitude to Mr. Arittra Bhattacharya for his kind help with drawing the phylogenetic tree.

